Stay Up to Date
Subscribe to our quarterly updates for all the latest MinEx CRC news
The isotopic signature of groundwater may be influenced by a number of processes, each of which may have different effect on the δ2H and δ18O values.
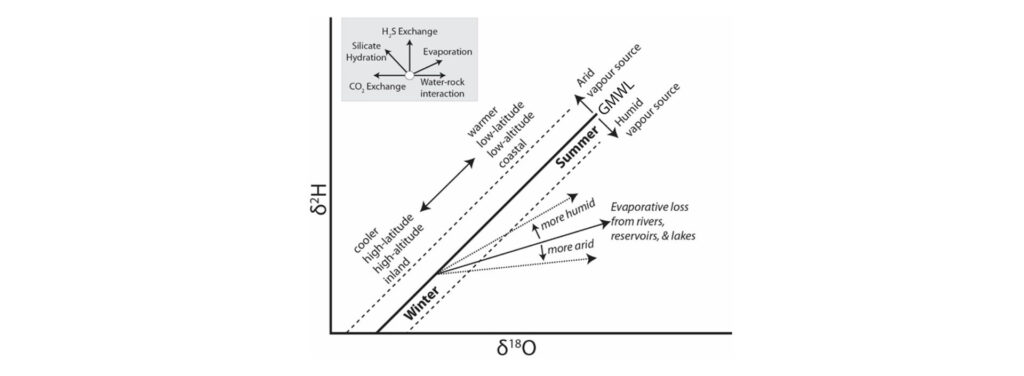
Figure 1: Summary figure showing the possible variation of O and H isotopes based on variation before, during and after precipitation.
The variation in the HO values highlights a trend for increasingly isotopically light meteoric waters as rainfall moves away from the coast. The range of sample points along the GMWL and the associated evaporation of these waters is therefore predominantly representative of the ‘continental effect’. Isotopically enriched rain forms and falls from a diminishing vapour mass, and residual vapour becomes isotopically depleted with respect to earlier rains from the same cloud resulting in meteoric waters with increasingly light δ18O and δ2H values the further from the coast precipitation occurs.
The O and H isotope results highlight potentially different water sources that may be related to faults, fractures or mineralisation. When combined with pathfinder chemistry and mineral saturation indices, O and H isotopes can be used to identify new areas of interest for mineral exploration.
Filtered water samples are sent to James Cook University (Cairns, Australia) for analysis of δ18O and δ2H by Cavity Ring-Down Spectrometry (CRDS). Water vapour was separated from non-volatile dissolved and particulate contaminants, minimising spectral interferences. Comparison of isotopic data for a range of water samples analysed by Diffusion Sampling-CRDS (DS-CRDS) and Isotope Ratio Mass Spectrometry shows significant linear correlations between the two methods allowing for accurate standardisation of DS-CRDS data. The isotopic effects of variable air temperature, water vapour concentration, water pumping rate and dissolved organic content were found to be either negligible or correctable by analysis of water standards.
It is well established that 34S/32S measurements may be useful in distinguishing rock sulfides from meteoric S. Lighter δ34S measured from SO4 are more commonly linked with the weathering of sulphides.
Sulfate in groundwater may be sourced from dissolution of sulfate minerals, precipitation, and dissolution of sulfides. Sulfate derived from the dissolution of sulfate minerals e.g. gypsum will possess heavier δ18O_SO4 values and variable, generally heavier δ34S, whilst data for the δ34S composition of precipitation in the region is partially dependant on the distance from the coast and the prevailing winds, with coastal regions gaining a higher proportion of 34S . Sulfides in the form of pyrite have been shown to react with water and dissolved molecular oxygen to form sulfate and Fe oxyhydroxides. Usher et al. (2004), have shown that water and not dissolved oxygen is the primary source of oxygen in the sulfate product. Similarly Heidel et al. (2009), suggested only a small proportion of molecular oxygen is incorporated into the sulfate during oxidation although this may increase for ultrafine pyrite.
Sulfur and oxygen isotopes from sulfate were analysed by adding excess BaCl2 to the sample to form a stable BaSO4 precipitate, which was subsequently separated by filtration in the laboratory, rinsed and dried. The BaSO4 precipitate was sent to the Isotope Science Laboratory, Department of Physics and Astronomy, University of Calgary (Canada), for δ34S and δ18O_SO4 isotope analysis on a Carlo Erba NA 1500 elemental analyser interfaced with a VG_ Prism II mass spectrometer. High temperature reaction of the BaSO4 with V2O5 and SiO2 generated SO2 for δ34S. Sulfur isotope compositions are reported using the conventional δ 34S scale in parts per thousand (‰).
Lead isotope composition within groundwater can potentially reflect mineralisation as low-temperature organic or inorganic processes do not fractionate Pb isotopes (Gulson and Mizon, 1979; Gulson, 1986; Caritat et al., 2005). Various mineralisation types have distinct Pb isotope signatures (Gulson et al., 1985; Gulson, 1986), allowing discrimination between various mineralisation types, or even between barren and economic sulfide accumulations.
Measurement of lead isotope ratios relies on the assumption that lead-rich (or uranium poor) phases preserve the lead isotopic signatures of their source U/Pb and Th/Pb ratios. These ratios are dependent both on the geological age due to the different rates of radioactive decay of 232Th to 208Pb, 235U to 207Pb and 238U to 206Pb, and on geological evolution as uranium and thorium are mobile elements during fractionation in crustal processes.
Borehole prospectivity can be deduced by comparing their Pb-Pb isotopic values to known radiogenic Pb domain of mineral deposits.
Lead isotope ratio analysis are performed with a newly installed state-of-the-art Agilent QQQ-8900 ICP-MS at QUT (Brisbane). Samples are injected into the plasma with a syringe-driven autosampler at 80 microliters per minute for 5 minutes, consuming 0.4 mL of solution. The mass spectrometer is tuned to yield ca. 1,000,000 counts per second (cps) per ppb analyte at a Ce-oxide formation rate of 0.8%. The mass spectrometer was run in no gas mode without mass shifts between the two quadrupoles. Masses 202 (Hg), 204 (Pb and Hg), 206 (Pb), 207 (Pb) and 208 (Pb) were analysed.
Dating of selected samples using C14 techniques shows minimum mixing ages for the groundwater. The age variability has some correlation between age and where the sample is located within the profile (higher elevation samples were older than those in the channels). Radiocarbon dating showing that waters are old (>5000 years) –indicates there is sufficient time for the waters to equilibrate with the surrounding rock and minerals. The younger waters were mostly restricted to samples closest to the topographical lows and/or were open bores where mixing from rainwater may have occurred.
14C samples are processed and analysed at ANSTO where the dissolved inorganic carbon (DIC) was converted into CO2 by acidifying the samples with H3PO4 and extracting the liberated CO2 gas using a cryogenic custom-built extraction line. The CO2 sample was then heated in a sealed glass tube, containing baked CuO, Ag wire and Cu wire, at 600 oC for 2 hours to remove any sulfur compounds that may have been liberated from the groundwater sample during CO2 extraction. The CO2 sample was then converted into graphite by reducing it with excess hydrogen gas in the presence of an iron catalyst at 600 0C.
The 14C contents were measured by accelerator mass spectrometry (AMS) using the ANSTO 1MV accelerator, VEGA. The 14C results were measured on 14C/12C ratios and reported as normalised percent Modern Carbon (pMC) with an average 1s error of 0.2 pMC. Radiocarbon analytical calculations were performed following the protocols outlined in Stuiver and Polach (1977). The uncorrected ages quoted are ‘conventional radiocarbon ages’ (years BP) and not calendar ages. This method assumes that atmospheric 14C levels have been constant in the past and uses a half-life of 5568 years. All 14C measurements were normalised against the oxalic acid international standard (HOxI). Isotopic fractionation normalisation of all sample activities are to the base of δ13C = -25‰, relative to PDB. The δ13C isotopic composition of the graphite AMS target was determined by Isotope Ratio Mass Spectrometry (IRMS) with a precision of 0.1‰, except for 3 samples where δ13C of the groundwater DIC was used instead (SJT 37, 46, 52). This graphite δ13C measurement is solely used as a correction in the calculations related to the radiocarbon measurement.
Workflows in case study areas that integrate this work is currently being produced.